What is Amyotrophic lateral sclerosis (ALS)?

 A health article about Amyotrophic lateral sclerosis (ALS) from Muscle Disorders the A to Z directory of
A health article about Amyotrophic lateral sclerosis (ALS) from Muscle Disorders the A to Z directory of
dealing with Health Problems & nutritional
Self Care Strategies
Often referred to as ALS, this is a disease that breaks down tissues in the nervous system (a neurodegenerative disease) of unknown cause that affects the nerves responsible for movement.
It is also known as motor neuron disease and Lou Gehrig's disease, after the baseball player whose career it ended.
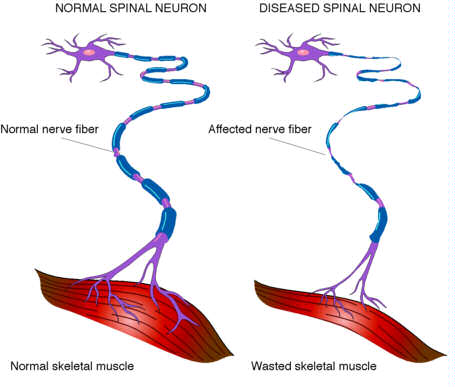
ALS is a disease of the motor neurons, those nerve cells reaching from the brain to the spinal cord (upper motor neurons) and the spinal cord to the peripheral nerves (lower motor neurons) that control muscle movement.
In Amyotrophic lateral sclerosis, for unknown reasons, these neurons die, leading to a progressive loss of the ability to move virtually any of the muscles in the body. ALS affects "voluntary" muscles, those controlled by conscious thought, such as the arm, leg, and trunk muscles.
Amyotrophic lateral sclerosis, in and of itself, does not affect sensation, thought processes, the heart muscle, or the "smooth" muscle of the digestive system, bladder, and other internal organs.
Most people with ALS retain function of their eye muscles as well. However, various forms of ALS may be associated with a loss of intellectual function (dementia) or sensory symptoms.
Amyotrophic lateral sclerosis progresses rapidly in most cases. It is fatal within three years for 50% of all people affected, and within five years for 80%. Ten percent of people with ALS live beyond eight years.
Causes of Amyotrophic lateral sclerosis
The symptoms of ALS are caused by the death of motor neurons in the spinal cord and brain. Normally, these neurons convey electrical messages from the brain to the muscles to stimulate movement in the arms, legs, trunk, neck, and head.
As motor neurons die, the muscles they enervate cannot be moved as effectively, and weakness results. In addition, lack of stimulation leads to muscle wasting, or loss of bulk.
Involvement of the upper motor neurons causes spasms and increased tone in the limbs, and abnormal reflexes. Involvement of the lower motor neurons causes muscle wasting and twitching (fasciculations).
Although many causes of motor neuron degeneration have been suggested for ALS, none has yet been proven responsible. Results of recent research have implicated toxic molecular fragments known as free radicals. Some evidence suggests that a cascade of events leads to excess free radical production inside motor neurons, leading to their death.
Why free radicals should be produced in excess amounts is unclear, as is whether this excess is the cause or the effect of other degenerative processes.
Additional agents within this toxic cascade may include excessive levels of a neurotransmitter known as glutamate, which may over-stimulate motor neurons, thereby increasing free-radical production, and a faulty detoxification enzyme known as SOD-1, for superoxide dismutase type 1.
The actual pathway of destruction is not known, however, nor is the trigger for the rapid degeneration that marks ALS. Further research may show that other pathways are involved, perhaps ones even more important than this one. Autoimmune factors or premature aging may play some role, as could viral agents or environmental toxins.
Two major forms of ALS are known: familial and sporadic. Familial ALS accounts for about 10% of all ALS cases. As the name suggests, familial ALS is believed to be caused by the inheritance of one or more faulty genes. About 15% of families with this type of ALS have mutations in the gene for SOD-1.
SOD-1 gene defects are dominant, meaning only one gene copy is needed to develop the disease. Therefore, a parent with the faulty gene has a 50% chance of passing the gene along to a child.
Sporadic ALS has no known cause. While many environmental toxins have been suggested as causes, to date no research has confirmed any of the candidates investigated, including aluminum and mercury and lead from dental fillings. As research progresses, it is likely that many cases of sporadic ALS will be shown to have a genetic basis as well.
A third type, called Western Pacific ALS, occurs in Guam and other Pacific islands. This form combines symptoms of both ALS and Parkinson's disease.
Symptoms of Amyotrophic lateral sclerosis
The earliest sign of ALS is most often weakness in the arms or legs, usually more pronounced on one side than the other at first. Loss of function is usually more rapid in the legs among people with familial ALS and in the arms among those with sporadic ALS.
Leg weakness may first become apparent by an increased frequency of stumbling on uneven pavement, or an unexplained difficulty climbing stairs. Arm weakness may lead to difficulty grasping and holding a cup, for instance, or loss of dexterity in the fingers.
Less often, the earliest sign of ALS is weakness in the bulbar muscles, those muscles in the mouth and throat that control chewing, swallowing, and speaking. A person with bulbar weakness may become hoarse or tired after speaking at length, or speech may become slurred.
In addition to weakness, the other cardinal signs of ALS are muscle wasting and persistent twitching (fasciculation). These are usually seen after weakness becomes obvious. Fasciculation is quite common in people without the disease, and is virtually never the first sign of ALS.
While initial weakness may be limited to one region, ALS almost always progresses rapidly to involve virtually all the voluntary muscle groups in the body. Later symptoms include loss of the ability to walk, to use the arms and hands, to speak clearly or at all, to swallow, and to hold the head up.
Weakness of the respiratory muscles makes breathing and coughing difficult, and poor swallowing control increases the likelihood of inhaling food or saliva (aspiration). Aspiration increases the likelihood of lung infection, which is often the cause of death.
With a ventilator and scrupulous bronchial hygiene, a person with ALS may live much longer than the average, although weakness and wasting will continue to erode any remaining functional abilities.
Most people with ALS continue to retain function of the extraocular muscles that move their eyes, allowing some communication to take place with simple blinks or through use of a computer-assisted device.
Diagnosis of Amyotrophic lateral sclerosis
The diagnosis of ALS begins with a complete medical history and physical exam, plus a neurological examination to determine the distribution and extent of weakness. An electrical test of muscle function, called an electromyogram, or EMG, is an important part of the diagnostic process.
Various other tests, including blood and urine tests, x rays, and CT scans, may be done to rule out other possible causes of the symptoms, such as tumors of the skull base or high cervical spinal cord, thyroid disease, spinal arthritis, lead poisoning, or severe vitamin deficiency. ALS is rarely misdiagnosed following a careful review of all these factors.
Treatment of Amyotrophic lateral sclerosis
There is no cure for ALS, and no treatment that can significantly alter its course. There are many things which can be done, however, to help maintain quality of life and to retain functional ability even in the face of progressive weakness.
As of the early 2000s, only one drug had been approved for treatment of ALS. Riluzole (Rilutek) appears to provide on average a three-month increase in life expectancy when taken regularly early in the disease, and shows a significant slowing of the loss of muscle strength. Riluzole acts by decreasing glutamate release from nerve terminals.
Experimental trials of nerve growth factor have not demonstrated any benefit. No other drug or vitamin currently available has been shown to have any effect on the course of ALS.
A physical therapist works with an affected person and family to implement exercise and stretching programs to maintain strength and range of motion, and to promote general health. Swimming may be a good choice for people with ALS, as it provides a low-impact workout to most muscle groups.
One result of chronic inactivity is contracture, or muscle shortening. Contractures limit a person's range of motion, and are often painful. Regular stretching can prevent contracture. Several drugs are available to reduce cramping, a common complaint in ALS.
An occupational therapist can help design solutions to movement and coordination problems, and provide advice on adaptive devices and home modifications.
Speech and swallowing difficulties can be minimized or delayed through training provided by a speech-language pathologist. This specialist can also provide advice on communication aids, including computer-assisted devices and simpler word boards.
Nutritional advice can be provided by a nutritionist. A person with ALS often needs softer foods to prevent jaw exhaustion or choking. Later in the disease, nutrition may be provided by a gastrostomy tube inserted into the stomach.
Mechanical ventilation may be used when breathing becomes too difficult. Modern mechanical ventilators are small and portable, allowing a person with ALS to maintain the maximum level of function and mobility.
Ventilation may be administered through a mouth or nose piece, or through a tracheostomy tube. This tube is inserted through a small hole made in the windpipe. In addition to providing direct access to the airway, the tube also decreases the risk aspiration.
While many people with rapidly progressing ALS choose not to use ventilators for lengthy periods, they are increasingly being used to prolong life for a short time.
The progressive nature of Amyotrophic lateral sclerosis means that most persons will eventually require full-time nursing care. This care is often provided by a spouse or other family member. While the skills involved are not difficult to learn, the physical and emotional burden of care can be overwhelming.
Caregivers need to recognize and provide for their own needs as well as those of people with ALS, to prevent depression, burnout, and bitterness.
Throughout the disease, a support group can provide important psychological aid to affected persons and their caregivers as they come to terms with the losses ALS inflicts. Support groups are sponsored by both the ALS Society and the Muscular Dystrophy Association.
Alternative treatment for Amyotrophic lateral sclerosis
Given the grave prognosis and absence of traditional medical treatments, it is not surprising that a large number of alternative treatments have been tried for ALS. Two studies published in 1988 suggested that amino-acid therapies may provide some improvement for some people with ALS.
While individual reports claim benefits for megavitamin therapy, herbal medicine, and removal of dental fillings, for instance, no evidence suggests that these offer any more than a brief psychological boost, often followed by a more severe letdown when it becomes apparent the disease has continued unabated. However, once the causes of ALS are bette
r understood, alternative therapies may be more intensively studied. For example, if damage by free radicals turns out to be the root of most of the symptoms, antioxidant vitamins and supplements may be used more routinely to slow the progression of ALS. Or, if environmental toxins are implicated, alternative therapies with the goal of detoxifying the body may be of some use.
Prognosis of Amyotrophic lateral sclerosis
ALS usually progresses rapidly, and leads to death from respiratory infection within three to five years in most cases. The slowest disease progression is seen in those who are young and have their first symptoms in the limbs. About 10% of people with ALS live longer than eight years.
Prevention Amyotrophic lateral sclerosis
Currently there is no known way to prevent ALS or to alter its course.
What are Muscle disorders?
Movement disorders are a group of diseases and syndromes affecting the ability to produce and control movement. Though it seems simple and effortless, normal movement in fact requires an astonishingly complex system of control.
Disruption of any portion of this system can cause a person to produce movements that are too weak, too forceful, too uncoordinated, or too poorly controlled for the task at hand. Unwanted movements may occur at rest.
Intentional movement may become impossible. Such conditions are called movement disorders.
(To read the rest of this article click on the Title above here.)

Free Report Reveals How to Get In Shape, Stay Active, And Start Living A Healthy Life While Balancing Your Work, Home And Family Needs!
Free Health Book.
See how easily you can fit simple, quick exercises into your daily life!
Click the link above or the book cover to get your free ebook & eCourse today!
|

to “Your Health Success”
our monthly F’R’E’E’ Newsletter
Back to Top of Muscle Amyotrophic lateral sclerosis (ALS) page
Subscribe to “Your Health Success” newsletter
Terms of Use |
Privacy Policy |
Disclaimer |
Site Map

============================================================================
SITE DISCLAIMER: Do these products “cure” anything? Of course not… but it stands to reason that if you cleanse your body and feed it the finest nutrition available, giving it everything it needs in balance, on a daily basis, that your body will do what nature intended, and give you the best possible chance to fend off sickness and disease. This Muscle Amyotrophic lateral sclerosis (ALS) information is not presented by a medical practitioner and is for educational and informational purposes only. The Muscle Amyotrophic lateral sclerosis (ALS) content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any Muscle Amyotrophic lateral sclerosis (ALS) questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read.
The Muscle Amyotrophic lateral sclerosis (ALS) resources on this site are not intended to be a substitute for professional advice. While all attempts have been made to verify Muscle Amyotrophic lateral sclerosis (ALS) information provided in this publication, neither the author nor the publisher assumes any responsibility for errors, omissions or contrary interpretation of the web site Muscle Amyotrophic lateral sclerosis (ALS) subject matter herein. The site Muscle Amyotrophic lateral sclerosis (ALS) contents are solely the opinion of the authors and should not be considered as a form of advice, direction and/or recommendation of any kind. If expert advice or counseling is needed, services of a competent professional should be sought. The author and the Publisher assume no responsibility or liability and specifically disclaim any warranty, express or implied for any Muscle Amyotrophic lateral sclerosis (ALS) products or services mentioned, or any techniques or Muscle Amyotrophic lateral sclerosis (ALS) practices described.
The purchaser or reader of thi Muscle Amyotrophic lateral sclerosis (ALS) s publication assumes responsibility for the use of these materials and information. Neither the author nor the Publisher assumes any responsibility or liability whatsoever on the behalf of any purchaser or reader of these Muscle Amyotrophic lateral sclerosis (ALS) materials. There is no guarantee of validity of accuracy. Any perceived slight of specific people or organizations is unintentional. This website and its creators are not responsible for the content of any sites linked to. Since natural and/or dietary supplements are not FDA approved they must be accompanied by a two-part disclaimer on the product label: that the statement has not been evaluated by FDA and that the product is not intended to "diagnose, treat, cure or prevent any disease."
============================================================================
Back to Top of page
